Band structure calculation with Qamuy¶
Install Qamuy Client SDK¶
If you are running this notebook on Google Colaboratory, then install Qamuy Client SDK and login to Qamuy by running the following 2 cells. Otherwise, run commands in the following 2 cells on a terminal since they require input from standard input, which cannot be handled on Jupyter Notebook.
[ ]:
!python -m pip install qamuy-client --extra-index-url https://download.qamuy.qunasys.com/simple/
Import Qamuy¶
[1]:
import qamuy.chemistry as qy
from qamuy.client import Client
from copy import deepcopy
import numpy as np
import matplotlib.pyplot as plt
# You can fill in your e-mail address and password.
client = Client(email_address="YOUR_EMAIL_ADDRESS", password="YOUR_PASSWORD")
input instance generation¶
[2]:
input = qy.QamuyChemistryInput()
ps = input.target_periodic_system
unit cell¶
[3]:
# see also https://pyscf.org/user/pbc/gto.html
BOHR = 0.529 # unit: angstrom
unitcell_x = 4. * BOHR
unitcell_yz = 1.
atoms = ["H", "H"]
coords = [[0., 0., 0.],[BOHR, 0., 0.]]
lattice_vec = [[unitcell_x, 0., 0.], [0., unitcell_yz, 0.], [0., 0., unitcell_yz]]
kpt_grid_shape = [1, 1, 1] # number of k-points in each direction.
dimension = 1
ps.geometry = qy.periodic_system_geometry(atoms, coords, lattice_vec, kpt_grid_shape)
ps.geometry.dimension = dimension
[4]:
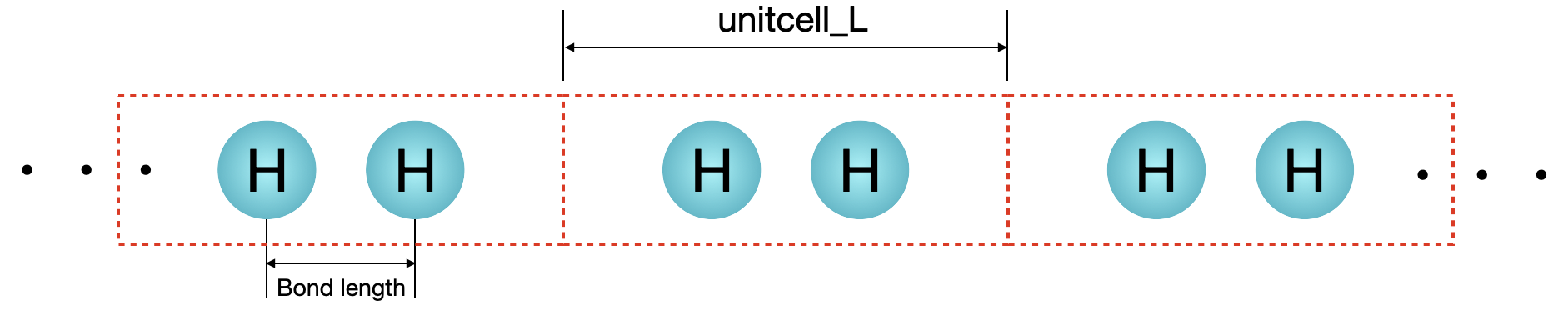
from PIL import Image
filename = "figs/hydrogen_chain.png"
im = Image.open(filename)
im
[4]:
basis, number of excited states, and multiplicity¶
[5]:
ps.basis = "sto-3g"
ps.num_excited_states = 0
ps.multiplicity = 1 # required (from ver. 0.18.1)
# CAS
ps.cas = qy.cas(2, 2)
solver, ansatz, optimizer, cost function, and quantum device¶
[6]:
input.solver.type = "VQE"
input.ansatz.type = "UCCD"
input.ansatz.is_state_real = True
input.ansatz.reference_state = "RHF"
input.ansatz.trotter_steps = 4
input.ansatz.initial_parameter = [0.0]
input.ansatz.init_param_random_seed = 1
input.optimizer.type = "BFGS"
input.optimizer.gtol = 1e-6
input.cost_function.type = "SIMPLE"
# exact simulator (state vector)
input.quantum_device.type = "EXACT_SIMULATOR"
properties to be evaluated¶
[7]:
properties = input.output_chemical_properties
properties.append(qy.output_chemical_property(target="band_structure", algorithm="QSE"))
properties.append(qy.output_chemical_property(target="band_structure", algorithm="QEOM"))
run¶
[8]:
kx_list = [round(0.05*i,2) for i in range(20)]
inputs = []
jobs = []
outputs = []
for kx in kx_list:
temp_input = deepcopy(input)
ps = temp_input.target_periodic_system
ps.scaled_center = [kx, 0., 0.]
inputs.append(temp_input)
for input_ in inputs:
jobs.append(client.submit(input_))
results = client.wait_and_get_job_results(jobs)
outputs = [result.output for result in results]
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 8 concurrent workers.
[Parallel(n_jobs=-1)]: Done 2 tasks | elapsed: 6.4min
[Parallel(n_jobs=-1)]: Done 8 out of 20 | elapsed: 6.5min remaining: 9.8min
[Parallel(n_jobs=-1)]: Done 11 out of 20 | elapsed: 6.5min remaining: 5.3min
[Parallel(n_jobs=-1)]: Done 14 out of 20 | elapsed: 6.5min remaining: 2.8min
[Parallel(n_jobs=-1)]: Done 17 out of 20 | elapsed: 6.7min remaining: 1.2min
[Parallel(n_jobs=-1)]: Done 20 out of 20 | elapsed: 6.8min remaining: 0.0s
[Parallel(n_jobs=-1)]: Done 20 out of 20 | elapsed: 6.8min finished
extract band energies¶
[9]:
kx_list = []
conduction_qse = []
conduction_qeom = []
conduction_hf = []
valence_qse = []
valence_qeom = []
valence_hf = []
for output in outputs:
q_result = output.molecule_result.quantum_device_result
gs_energy = q_result.evaluated_properties[0].energy.values[0].value
band_energies_qse = qy.get_evaluated_properties(q_result, "band_structure")[0].values
band_energies_qeom = qy.get_evaluated_properties(q_result, "band_structure")[1].values
hf_result = output.molecule_result.hf_result
kx_list.append(band_energies_qse[0].kpt[0])
conduction = []
valence = []
for band_dict in band_energies_qse:
if band_dict.band == "conduction band":
conduction.append(band_dict.value)
elif band_dict.band == "valence band":
valence.append(band_dict.value)
idx_conduction = np.abs(np.asarray(conduction) - gs_energy).argmin()
idx_valence = np.abs(np.asarray(valence) - gs_energy).argmin()
conduction_qse.append(conduction[idx_conduction])
valence_qse.append(valence[idx_valence])
conduction = []
valence = []
for band_dict in band_energies_qeom:
if band_dict.band == "conduction band":
conduction.append(band_dict.value)
elif band_dict.band == "valence band":
valence.append(band_dict.value)
idx_conduction = np.abs(np.asarray(conduction) - gs_energy).argmin()
idx_valence = np.abs(np.asarray(valence) - gs_energy).argmin()
conduction_qeom.append(conduction[idx_conduction])
valence_qeom.append(valence[idx_valence])
conduction_hf.append(hf_result.mo_energy[1])
valence_hf.append(hf_result.mo_energy[0])
plot¶
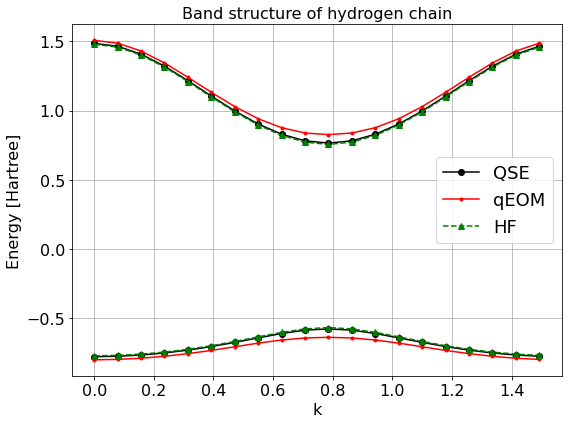
[10]:
fig = plt.figure(figsize=(8, 6))
ax = fig.add_subplot(111)
ax.plot(kx_list, conduction_qse, color="black", marker="o", label="QSE")
ax.plot(kx_list, conduction_qeom, color="red", marker=".", label="qEOM")
ax.plot(kx_list, conduction_hf, ls="dashed", marker="^", color="green", label="HF")
ax.plot(kx_list, valence_qse, marker="o", color="black")
ax.plot(kx_list, valence_qeom, marker=".", color="red")
ax.plot(kx_list, valence_hf, marker="^", ls="dashed", color="green")
ax.grid(True, which='both', axis='both')
ax.set_xlabel("k")
ax.set_ylabel("Energy [Hartree]")
ax.set_title("Band structure of hydrogen chain")
ax.legend(fontsize=18)
for item in ([ax.title, ax.xaxis.label, ax.yaxis.label] + ax.get_xticklabels() + ax.get_yticklabels()):
item.set_fontsize(16)
fig.tight_layout()