Molecular dynamics simulation with Qamuy¶
Install Qamuy Client SDK¶
If you are running this notebook on Google Colaboratory, then install Qamuy Client SDK and login to Qamuy by running the following 2 cells. Otherwise, run commands in the following 2 cells on a terminal since they require input from standard input, which cannot be handled on Jupyter Notebook.
[ ]:
!python -m pip install qamuy-client --extra-index-url https://download.qamuy.qunasys.com/simple/
Define Methods for MD¶
[1]:
import os
from functools import reduce
import numpy as np
import argparse
import yaml
import matplotlib.pyplot as plt
from qamuy.client import Client
from qamuy.utils.file_io import save, load_input
# You can fill in your e-mail address and password.
client = Client(email_address="YOUR_EMAIL_ADDRESS", password="YOUR_PASSWORD")
ao = 5.2917721067e-11 # bohr
NA = 6.022140857e23 # mol-1
me = 9.10938356e-31 # kg / a. u.
hbarOEh = 2.418884326509e-17 # s / a. u.
kt = hbarOEh * 1.0e15 # a.u. to fs
kr = ao * 1.0e10 # a.u. to A
mass_list = {
"H": 1.00782504,
"C": 12.00000000,
"N": 14.00307401,
"O": 15.99491464,
"F": 18.9984032,
"Cl": 35.453,
}
mass_list_au = {key: val * (1.0e-3 / NA / me) for key, val in mass_list.items()}
folder = "MD/"
temp_folder = folder + "TEMP/"
output_folder = folder + "OUTPUT/"
qamuy_setting_file = folder + "qamuy_setting.yaml"
qamuy_input_file = temp_folder + "qamuy_input.yaml"
qamuy_output_file = temp_folder + "qamuy_output.yaml"
output_file = output_folder + "output.log"
output_energy_file = output_folder + "energy.log"
output_xyz_file = output_folder + "geometry.xyz"
output_vel_file = output_folder + "velocity.log"
init_xyz_file = folder + "init.xyz"
init_vel_file = folder + "init.vel"
class Setting:
def __init__(self, time_step, time_limit, state, classical=False):
self.time_step = time_step / kt
self.time_limit = time_limit / kt
self.qamuy_setting = self.read_qamuy_setting()
self.classical = classical
self.client = client
self.state = state
def read_qamuy_setting(self) -> dict:
with open(qamuy_setting_file, "r") as f:
return yaml.safe_load(f)
def write_qamuy_input(self):
with open(qamuy_input_file, "w") as f:
yaml.dump(self.qamuy_setting, f, indent=2)
def read_qamuy_output(self):
with open(qamuy_output_file, "r") as f:
output = yaml.safe_load(f)
if self.classical:
results = output["molecule_result"]["post_hf_results"][0]
opt_params = []
else:
results = output["molecule_result"]["quantum_device_result"]
opt_params = results["vqe_log"]["opt_params"]
properies = results["evaluated_properties"]
ene = properies[0]["energy"]["values"][self.state]["value"]
grad = properies[4]["gradient"]["values"][0]["value"]
qamuy_log = Qamuy_log(
opt_params=opt_params, potential=ene, force=-np.array(grad)
)
return qamuy_log
def run_qamuy(self, step):
atoms = step.atoms
rr = step.position
geometry = self.qamuy_setting["target_molecule"]["geometry"]
geometry["atoms"] = atoms
geometry["coordinates"] = [(rr[3 * i : 3 * i + 3] * kr).tolist() for i in range(len(atoms))]
self.qamuy_setting["ansatz"]["initial_parameter"] = (
step.qamuy_log.opt_params if step.qamuy_log else []
)
self.write_qamuy_input()
input_pb = load_input(qamuy_input_file)
job = self.client.submit(input_pb)
output_pb = self.client.wait_and_get_job_results([job])[0]
save(output_pb.output, filename=qamuy_output_file)
return self.read_qamuy_output()
class Qamuy_log:
def __init__(self, opt_params, potential, force):
self.opt_params = opt_params
self.potential = potential
self.force = force
class Step:
def __init__(
self, atoms, velocity, position, time=0.0, filename="output", qamuy_log=None
):
self.filename = filename
self.atoms = atoms
self.time = time
self.velocity = velocity
self.position = position
self.qamuy_log = qamuy_log
self.mass = np.array(
reduce(lambda ss, x: ss + [mass_list_au[x]] * 3, atoms, [])
)
def cal_PE(self) -> float:
return self.qamuy_log.potential
def cal_KE(self) -> float:
v = self.velocity
return 0.5 * sum(step.mass * v * v)
def cal_totE(self) -> float:
return self.cal_PE() + self.cal_KE()
def velocity_verlet(setting: Setting, step: Step) -> Step:
# all terms in a.u.
dt = setting.time_step
r = step.position
v = step.velocity
F = step.qamuy_log.force
m = step.mass
step.time = step.time + dt
step.position = r + v * dt + 0.5 * dt * dt / m * F
q_log = setting.run_qamuy(step)
if q_log is not None:
new_F = q_log.force
step.velocity = v + 0.5 * dt / step.mass * (F + new_F)
step.qamuy_log = q_log
return step
def run_MD(setting: Setting, step: Step):
os.makedirs(temp_folder, exist_ok=True)
os.makedirs(output_folder, exist_ok=True)
step.qamuy_log = setting.run_qamuy(step)
dt = setting.time_step
N = int((setting.time_limit - step.time) / dt)
write_outputs(step, "w")
for i in range(N):
step = velocity_verlet(setting, step)
write_outputs(step, "a")
def write_outputs(step, mode):
atoms = step.atoms
t = step.time
length = len(atoms)
# write xyz
header = "%2d\n% 3.15e %5.6f\n" % (length, step.qamuy_log.potential, t * kt)
dd = reduce(
lambda ss, i: ss
+ "%2s % 3.15e % 3.15e % 3.15e\n"
% ((atoms[i],) + tuple(step.position[3 * i : 3 * i + 3] * kr)),
range(length),
header,
)
with open(output_xyz_file, mode) as f:
f.write(dd)
# write velocity
dd = reduce(
lambda ss, i: ss
+ "%2s % 3.15e % 3.15e % 3.15e\n"
% ((atoms[i],) + tuple(step.velocity[3 * i : 3 * i + 3])),
range(length),
header,
)
with open(output_vel_file, mode) as f:
f.write(dd)
# write energy
if mode == "w":
with open(output_energy_file, mode) as f:
f.write(
"# %7s %19s %19s %19s\n"
% ("Time", "Potential Eenergy", "Kinetic Eenergy", "Total Eenergy")
)
with open(output_energy_file, "a") as f:
f.write(
"% 5.6f % 3.15e % 3.15e % 3.15e\n"
% (t * kt, step.cal_PE(), step.cal_KE(), step.cal_totE())
)
def read_initial_file():
with open(init_xyz_file, "r") as f:
data = f.read()
lines = data.split("\n")
n = int(lines[0])
atoms, coords = zip(
*[(l.split()[0], [float(x) for x in l.split()[1:4]]) for l in lines[2 : n + 2]]
)
with open(init_vel_file, "r") as f:
data = f.read()
vels = np.array([float(x) for x in data.split()])
return list(atoms), (np.array(coords).ravel() / kr), vels
Setting for VQE¶
[2]:
os.makedirs(folder, exist_ok=True)
setting = (
"""target_molecule:
geometry:
atoms: []
coordinates: []
basis: 6-31g
cas:
active_orb: 2
active_ele: 2
charge: 0
multiplicity: 1
num_excited_states: 0
# Set Chemical Properties to Be Calculated
output_chemical_properties:
- gradient:
type: ANALYTICAL_PYSCF
parameters:
- state: 0
# Set VQE Solver Type
solver:
type: VQE
# Set Qubit Mapping
mapping:
type: JORDAN_WIGNER
# Set Cost Function
cost_function:
type: SIMPLE
particle_number_weight: 10.0
s2_number_weight: 10.0
sz_number_weight: 10.0
# Set Ansatz
ansatz:
type: SYMMETRY_PRESERVING
initial_parameter: []
use_random_initial_guess: true
is_state_real: true
reference_state: RHF
depth: 5
# Set Optimizer
optimizer:
type: BFGS
ftol: 1.0e-06
gtol: 1.0e-06
max_fev: 100000
max_iter: 100000
max_run: 100000
"""
)
with open(qamuy_setting_file, "w") as f:
f.write(setting)
Generate initial nulcear coodinates and velocities¶
[3]:
# coordinates in Å
xyz = (
"""3
H -1.5 -1.5 0.0
O 0.0 0.0 0.0
H 1.5 -1.5 0.0
"""
)
with open(init_xyz_file, "w") as f:
f.write(xyz)
# velocities in atomic unit
vel = (
"""0.005 0.0 0.0
0.0 0.0 0.0
-0.005 0.0 0.0
"""
)
with open(init_vel_file, "w") as f:
f.write(vel)
[4]:
import py3Dmol
view = py3Dmol.view(width=400, height=300)
view.addModel(xyz, "xyz")
view.setStyle({"sphere": {}})
view.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
Setting for MD¶
[5]:
time_step = 0.5 # fs
time_limit = 5.0 # fs
init_state = 0 # ground state
setting = Setting(
time_step=time_step,
time_limit=time_limit,
state=init_state,
)
Run MD¶
[6]:
atoms, coords, vels = read_initial_file()
step = Step(atoms=atoms, position=coords, velocity=vels)
run_MD(setting, step)
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 27.6s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 27.6s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 21.7s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 21.7s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 16.3s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 16.3s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 21.3s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 21.3s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 21.3s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 21.3s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 10.9s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 11.0s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 11.0s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 11.0s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 11.1s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 11.1s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 16.3s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 16.3s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 21.3s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 21.3s finished
[Parallel(n_jobs=-1)]: Using backend ThreadingBackend with 4 concurrent workers.
[Parallel(n_jobs=-1)]: Done 1 tasks | elapsed: 26.8s
[Parallel(n_jobs=-1)]: Done 1 out of 1 | elapsed: 26.8s finished
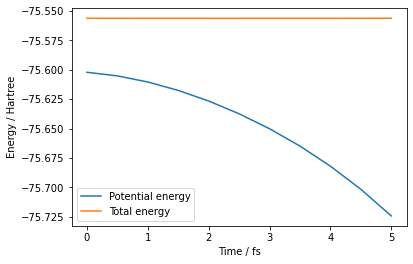
Plot Results¶
[7]:
def plot_energy(ff):
es = np.loadtxt(ff, comments="#").T
plt.plot(es[0], es[1], label="Potential energy")
plt.plot(es[0], es[3], label="Total energy")
plt.xlabel("Time / fs")
plt.ylabel("Energy / Hartree")
plt.legend()
plt.show()
plot_energy(output_energy_file)
[8]:
with open(output_xyz_file) as f:
output_xyz = f.read()
view = py3Dmol.view(width=400, height=300)
view.addModelsAsFrames(output_xyz, "xyz")
view.setStyle({"sphere": {}})
view.animate({'loop': "forward"})
view.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol